Importing and manipulating data for TRANSPIRE analysis¶
The backbone of analysis with TRANSPIRE relies heavily on Pandas DataFrames with multi-level indicies. This provides a powerful and flexible framework for data manipulation. To make this analysis more accessible to those without a strong background or familiarity with Pandas, TRANSPIRE provides utilities to simplify data loading and manipulation. For more advanced users, they may prefer to forego the built-in TRANSPIRE utilities for their own functions.
[47]:
import TRANSPIRE
1. Define file path and load data¶
[48]:
f = 'mydata_no_localizations.csv'
df = TRANSPIRE.data.import_data.load_data(f)
df
[48]:
| condition | uninfected | infected | |||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| fraction | 1 | 2 | 3 | 4 | 5 | 6 | 1 | 2 | 3 | 4 | 5 | 6 | |
| accession | gene name | ||||||||||||
| A0AVT1 | UBA6 | 0.342416 | 0.346408 | 0.147271 | 0.090486 | 0.043105 | 0.030314 | 0.056764 | 0.095989 | 0.094597 | 0.139616 | 0.345400 | 0.257444 |
| A0FGR8 | ESYT2 | 0.200266 | 0.288014 | 0.264795 | 0.152079 | 0.061482 | 0.020638 | 0.141740 | 0.250193 | 0.250726 | 0.142513 | 0.153470 | 0.049585 |
| A1L0T0 | ILVBL | 0.193194 | 0.333657 | 0.239388 | 0.160094 | 0.065503 | 0.018098 | 0.127224 | 0.265472 | 0.228027 | 0.143158 | 0.173794 | 0.054383 |
| A1L188 | NDUFAF8 | NaN | NaN | NaN | NaN | NaN | NaN | 0.034472 | 0.063834 | 0.198874 | 0.395500 | 0.214612 | 0.092709 |
| A2RRP1 | NBAS | 0.266384 | 0.305499 | 0.207475 | 0.134155 | 0.052810 | 0.019836 | 0.155677 | 0.248836 | 0.210393 | 0.128968 | 0.163133 | 0.082520 |
| ... | ... | ... | ... | ... | ... | ... | ... | ... | ... | ... | ... | ... | ... |
| Q9Y6Q2 | STON1 | 0.468515 | 0.291495 | 0.107717 | 0.079150 | 0.037682 | 0.015441 | NaN | NaN | NaN | NaN | NaN | NaN |
| Q9Y6R0 | NUMBL | 0.281558 | 0.325676 | 0.221316 | 0.099426 | 0.049386 | 0.022638 | NaN | NaN | NaN | NaN | NaN | NaN |
| Q9Y6R1 | SLC4A4 | 0.319321 | 0.299931 | 0.208633 | 0.104363 | 0.042091 | 0.017095 | NaN | NaN | NaN | NaN | NaN | NaN |
| Q9Y6V0 | PCLO | 0.289078 | 0.369018 | 0.110457 | 0.088003 | 0.097068 | 0.014004 | 0.204732 | 0.314821 | 0.167343 | 0.128688 | 0.131086 | 0.053329 |
| Q9Y6W5 | WASF2 | 0.483214 | 0.298740 | 0.120254 | 0.054733 | 0.034164 | 0.014242 | NaN | NaN | NaN | NaN | NaN | NaN |
3304 rows × 12 columns
Note that the DataFrame above does not contain a “localization” index level. Thus, we need to assign a set of organelle markers to the data in order to run the TRANSPIRE analysis.
2. Add subcellular localization annotation to markers (optional)¶
[49]:
df_with_markers = TRANSPIRE.data.import_data.add_markers(df, 'human_fibroblast')
df_with_markers
[49]:
| condition | uninfected | infected | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| fraction | 1 | 2 | 3 | 4 | 5 | 6 | 1 | 2 | 3 | 4 | 5 | 6 | ||
| accession | gene name | localization | ||||||||||||
| A0AVT1 | UBA6 | NaN | 0.342416 | 0.346408 | 0.147271 | 0.090486 | 0.043105 | 0.030314 | 0.056764 | 0.095989 | 0.094597 | 0.139616 | 0.345400 | 0.257444 |
| A0FGR8 | ESYT2 | NaN | 0.200266 | 0.288014 | 0.264795 | 0.152079 | 0.061482 | 0.020638 | 0.141740 | 0.250193 | 0.250726 | 0.142513 | 0.153470 | 0.049585 |
| A1L0T0 | ILVBL | NaN | 0.193194 | 0.333657 | 0.239388 | 0.160094 | 0.065503 | 0.018098 | 0.127224 | 0.265472 | 0.228027 | 0.143158 | 0.173794 | 0.054383 |
| A1L188 | NDUFAF8 | NaN | NaN | NaN | NaN | NaN | NaN | NaN | 0.034472 | 0.063834 | 0.198874 | 0.395500 | 0.214612 | 0.092709 |
| A2RRP1 | NBAS | NaN | 0.266384 | 0.305499 | 0.207475 | 0.134155 | 0.052810 | 0.019836 | 0.155677 | 0.248836 | 0.210393 | 0.128968 | 0.163133 | 0.082520 |
| ... | ... | ... | ... | ... | ... | ... | ... | ... | ... | ... | ... | ... | ... | ... |
| Q9Y6Q2 | STON1 | Cytosol | 0.468515 | 0.291495 | 0.107717 | 0.079150 | 0.037682 | 0.015441 | NaN | NaN | NaN | NaN | NaN | NaN |
| Q9Y6R0 | NUMBL | NaN | 0.281558 | 0.325676 | 0.221316 | 0.099426 | 0.049386 | 0.022638 | NaN | NaN | NaN | NaN | NaN | NaN |
| Q9Y6R1 | SLC4A4 | NaN | 0.319321 | 0.299931 | 0.208633 | 0.104363 | 0.042091 | 0.017095 | NaN | NaN | NaN | NaN | NaN | NaN |
| Q9Y6V0 | PCLO | NaN | 0.289078 | 0.369018 | 0.110457 | 0.088003 | 0.097068 | 0.014004 | 0.204732 | 0.314821 | 0.167343 | 0.128688 | 0.131086 | 0.053329 |
| Q9Y6W5 | WASF2 | Cytosol | 0.483214 | 0.298740 | 0.120254 | 0.054733 | 0.034164 | 0.014242 | NaN | NaN | NaN | NaN | NaN | NaN |
3304 rows × 12 columns
Notice how this DataFrame now has a “localization” index level. At this point, we should visualize our data and see how well-separated are organelle marker classes are to determine if we should group any together (e.g. for less-complex fractionation workflows).
[35]:
# select the subset of the dataframe with non-null localization values (i.e. the markers)
marker_profiles = df_with_markers[df_with_markers.index.get_level_values('localization').notnull()]
# pivot the dataframe so that the "condition" column level is now an index level
marker_profiles_pivot = marker_profiles.stack('condition')
# "melt" the dataframe so that the fractions across the top of the dataframe
# become their own column of values, associated with a corresponding relative abundance value
markers_to_plot = marker_profiles_pivot.reset_index().melt(marker_profiles_pivot.index.names)
markers_to_plot
[35]:
| accession | gene name | localization | condition | fraction | value | |
|---|---|---|---|---|---|---|
| 0 | O00115 | DNASE2 | Lysosome | infected | 1 | 0.091988 |
| 1 | O00115 | DNASE2 | Lysosome | uninfected | 1 | 0.121081 |
| 2 | O00116 | AGPS | Peroxisome | infected | 1 | 0.035467 |
| 3 | O00116 | AGPS | Peroxisome | uninfected | 1 | 0.066992 |
| 4 | O00151 | PDLIM1 | Cytosol | infected | 1 | 0.298505 |
| ... | ... | ... | ... | ... | ... | ... |
| 5743 | Q9Y6G9 | DYNC1LI1 | Cytosol | uninfected | 6 | 0.030355 |
| 5744 | Q9Y6N5 | SQOR | Mitochondria | infected | 6 | 0.127455 |
| 5745 | Q9Y6N5 | SQOR | Mitochondria | uninfected | 6 | 0.049427 |
| 5746 | Q9Y6Q2 | STON1 | Cytosol | uninfected | 6 | 0.015441 |
| 5747 | Q9Y6W5 | WASF2 | Cytosol | uninfected | 6 | 0.014242 |
5748 rows × 6 columns
*Now, lets look at the marker protein profiles*
[36]:
import seaborn as sns
import matplotlib.pyplot as plt
g = sns.FacetGrid(data = markers_to_plot, col = 'condition', hue='localization')
g.map(sns.lineplot, 'fraction','value')
g.add_legend()
plt.show()
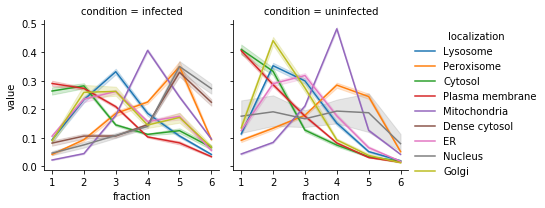
In this data, we can see that the plasma membrane and cytosol have overlapping profiles. Same with the ER, Lysosome, and Golgi, as well as the Dense cytosol and nucleus. If we desire, TRANSPIRE has a built in utility to group organlles together. Note that organelles excluded from the mapping defined below will retain their original assignments
[51]:
organelle_mapping = {
'ER': 'ER/Golgi/Lysosome',
'Golgi': 'ER/Golgi/Lysosome',
'Lysosome': 'ER/Golgi/Lysosome',
'Plasma membrane':'PM/Cytosol',
'Cytosol': 'PM/Cytosol',
'Dense cytosol': 'DC/Nucleus',
'Nucleus': 'DC/Nucleus'
}
df_grouped = TRANSPIRE.utils.group_organelles(df_with_markers, organelle_mapping)
Now if we plot the profiles of the grouped markers, we can see that they are much more clearly separated¶
[38]:
g = sns.FacetGrid(data = df_grouped.stack('condition').reset_index().melt(df_grouped.index.names+['condition']),
col = 'condition', hue='localization')
g.map(sns.lineplot, 'fraction','value')
g.add_legend()
plt.show()
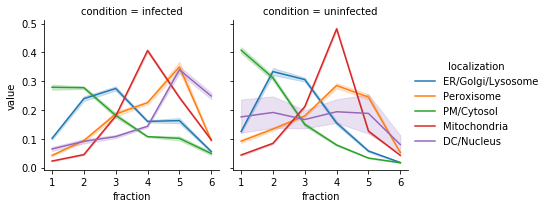
3. Define comparisons to make¶
Now we need to define which conditions we want to make comparisons between. In this dataset, we only have infected vs. uninfected conditions. Note that these comparisons MUST match the names of your condition columns–otherwise TRANSPIRE will fail
[39]:
comparisons = [('uninfected', 'infected')]
4. Generate synthetic translocations and label encodings¶
[41]:
synthetic_translocations = TRANSPIRE.data.generate_translocations.make_translocations(df_grouped, comparisons)
synthetic_translocations
[41]:
| 1 | 2 | 3 | 4 | 5 | 6 | 7 | 8 | 9 | 10 | 11 | 12 | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| accession_A | gene name_A | localization_A | condition_A | accession_B | gene name_B | localization_B | condition_B | label | ||||||||||||
| O00115 | DNASE2 | ER/Golgi/Lysosome | uninfected | O00115 | DNASE2 | ER/Golgi/Lysosome | infected | ER/Golgi/Lysosome to ER/Golgi/Lysosome | 0.121081 | 0.378059 | 0.287889 | 0.142575 | 0.056295 | 0.014235 | 0.091988 | 0.249011 | 0.334238 | 0.181254 | 0.104927 | 0.041090 |
| O00116 | AGPS | Peroxisome | infected | ER/Golgi/Lysosome to Peroxisome | 0.121081 | 0.378059 | 0.287889 | 0.142575 | 0.056295 | 0.014235 | 0.035467 | 0.087956 | 0.190615 | 0.228672 | 0.353767 | 0.095804 | ||||
| O00151 | PDLIM1 | PM/Cytosol | infected | ER/Golgi/Lysosome to PM/Cytosol | 0.121081 | 0.378059 | 0.287889 | 0.142575 | 0.056295 | 0.014235 | 0.298505 | 0.273591 | 0.136350 | 0.124446 | 0.101276 | 0.051836 | ||||
| O00161 | SNAP23 | PM/Cytosol | infected | ER/Golgi/Lysosome to PM/Cytosol | 0.121081 | 0.378059 | 0.287889 | 0.142575 | 0.056295 | 0.014235 | 0.303401 | 0.275456 | 0.219409 | 0.094259 | 0.065571 | 0.027320 | ||||
| O00186 | STXBP3 | PM/Cytosol | infected | ER/Golgi/Lysosome to PM/Cytosol | 0.121081 | 0.378059 | 0.287889 | 0.142575 | 0.056295 | 0.014235 | 0.251250 | 0.279922 | 0.220812 | 0.102742 | 0.089897 | 0.047200 | ||||
| ... | ... | ... | ... | ... | ... | ... | ... | ... | ... | ... | ... | ... | ... | ... | ... | ... | ... | ... | ... | ... |
| Q9Y6N5 | SQOR | Mitochondria | uninfected | Q9Y673 | ALG5 | ER/Golgi/Lysosome | infected | Mitochondria to ER/Golgi/Lysosome | 0.035159 | 0.085702 | 0.215704 | 0.471670 | 0.115302 | 0.049427 | 0.141876 | 0.246472 | 0.287558 | 0.145362 | 0.118529 | 0.041669 |
| Q9Y676 | MRPS18B | Mitochondria | infected | Mitochondria to Mitochondria | 0.035159 | 0.085702 | 0.215704 | 0.471670 | 0.115302 | 0.049427 | 0.016078 | 0.036276 | 0.165740 | 0.417482 | 0.257843 | 0.093664 | ||||
| Q9Y680 | FKBP7 | ER/Golgi/Lysosome | infected | Mitochondria to ER/Golgi/Lysosome | 0.035159 | 0.085702 | 0.215704 | 0.471670 | 0.115302 | 0.049427 | 0.129000 | 0.285763 | 0.290694 | 0.140269 | 0.108666 | 0.037075 | ||||
| Q9Y6G3 | MRPL42 | Mitochondria | infected | Mitochondria to Mitochondria | 0.035159 | 0.085702 | 0.215704 | 0.471670 | 0.115302 | 0.049427 | 0.022533 | 0.052818 | 0.178052 | 0.399956 | 0.237964 | 0.098262 | ||||
| Q9Y6N5 | SQOR | Mitochondria | infected | Mitochondria to Mitochondria | 0.035159 | 0.085702 | 0.215704 | 0.471670 | 0.115302 | 0.049427 | 0.023746 | 0.042218 | 0.201027 | 0.353196 | 0.246338 | 0.127455 |
147456 rows × 12 columns
[44]:
# generate mappings to encode the labels as integers for model training
mapping, mapping_r = TRANSPIRE.utils.get_mapping(df_grouped)
mapping # for encoding labels
[44]:
DC/Nucleus to DC/Nucleus 0
DC/Nucleus to ER/Golgi/Lysosome 1
DC/Nucleus to Mitochondria 2
DC/Nucleus to PM/Cytosol 3
DC/Nucleus to Peroxisome 4
ER/Golgi/Lysosome to DC/Nucleus 5
ER/Golgi/Lysosome to ER/Golgi/Lysosome 6
ER/Golgi/Lysosome to Mitochondria 7
ER/Golgi/Lysosome to PM/Cytosol 8
ER/Golgi/Lysosome to Peroxisome 9
Mitochondria to DC/Nucleus 10
Mitochondria to ER/Golgi/Lysosome 11
Mitochondria to Mitochondria 12
Mitochondria to PM/Cytosol 13
Mitochondria to Peroxisome 14
PM/Cytosol to DC/Nucleus 15
PM/Cytosol to ER/Golgi/Lysosome 16
PM/Cytosol to Mitochondria 17
PM/Cytosol to PM/Cytosol 18
PM/Cytosol to Peroxisome 19
Peroxisome to DC/Nucleus 20
Peroxisome to ER/Golgi/Lysosome 21
Peroxisome to Mitochondria 22
Peroxisome to PM/Cytosol 23
Peroxisome to Peroxisome 24
dtype: int64
[45]:
mapping_r # for de-encoding labels
[45]:
0 DC/Nucleus to DC/Nucleus
1 DC/Nucleus to ER/Golgi/Lysosome
2 DC/Nucleus to Mitochondria
3 DC/Nucleus to PM/Cytosol
4 DC/Nucleus to Peroxisome
5 ER/Golgi/Lysosome to DC/Nucleus
6 ER/Golgi/Lysosome to ER/Golgi/Lysosome
7 ER/Golgi/Lysosome to Mitochondria
8 ER/Golgi/Lysosome to PM/Cytosol
9 ER/Golgi/Lysosome to Peroxisome
10 Mitochondria to DC/Nucleus
11 Mitochondria to ER/Golgi/Lysosome
12 Mitochondria to Mitochondria
13 Mitochondria to PM/Cytosol
14 Mitochondria to Peroxisome
15 PM/Cytosol to DC/Nucleus
16 PM/Cytosol to ER/Golgi/Lysosome
17 PM/Cytosol to Mitochondria
18 PM/Cytosol to PM/Cytosol
19 PM/Cytosol to Peroxisome
20 Peroxisome to DC/Nucleus
21 Peroxisome to ER/Golgi/Lysosome
22 Peroxisome to Mitochondria
23 Peroxisome to PM/Cytosol
24 Peroxisome to Peroxisome
dtype: object
Now we have generated the synthetic translocation profiles that TRANSPIRE requires for model training and analysis. See the next notebook on hyperparameter optimization or skip to the notebook on final model fitting and evaluation to learn how to build and fit models for subsequent analysis